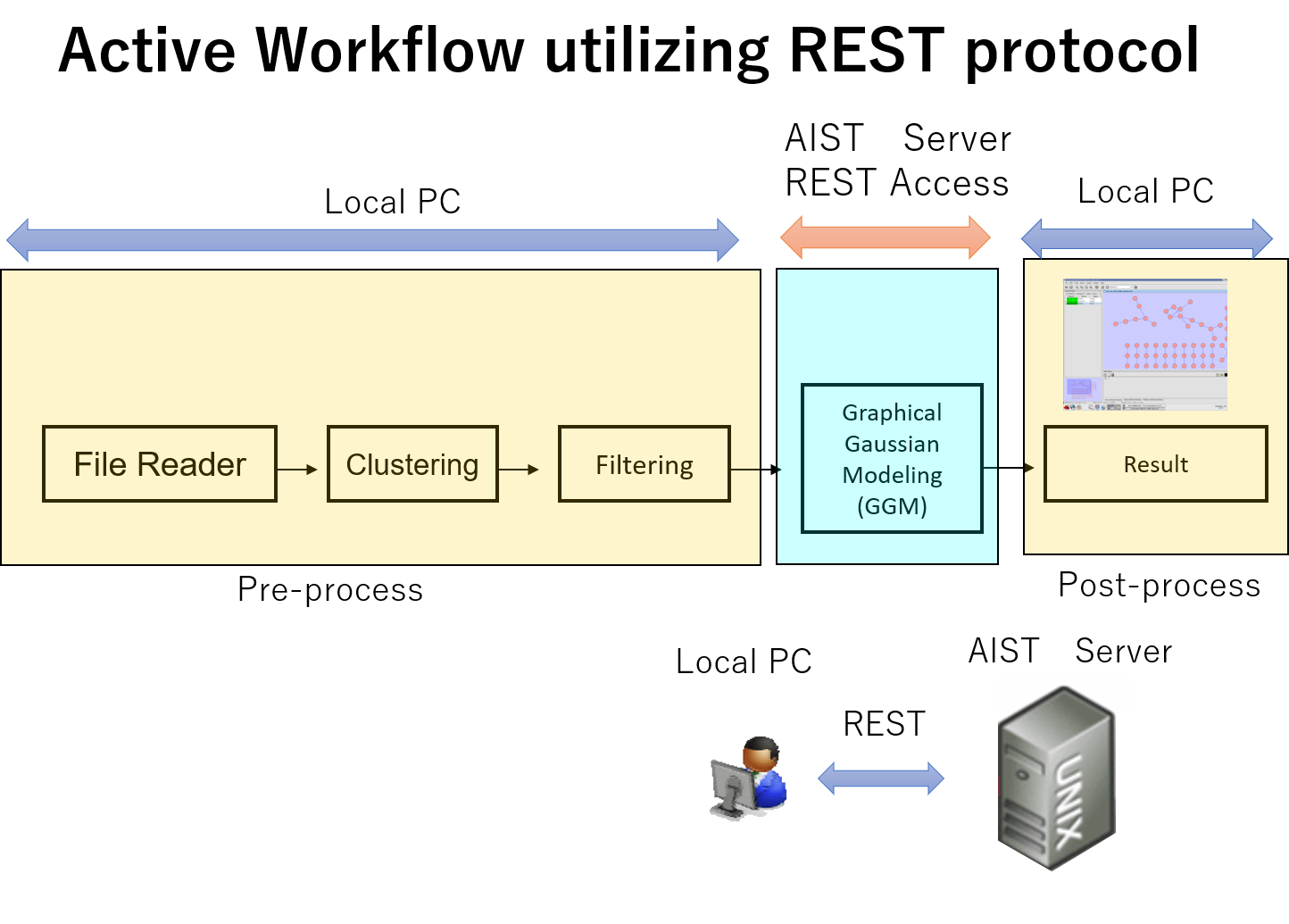
ACTIVE WORKFLOW
The user can select workflows that he or she wishes, using workflow platform (KNIME), on a client PC or a server. Within the workflow platform various components or parameters can be specified.
We use a free version of KNIME developed at University of Konstanz. KNIME is an eclipse-based workflow platform and uses nodes as processing units. Users can construct workflows, read data, calculate, analyze and visualize by combining those nodes. We also develop dynamic analysis platforms using semantic web technologies.
Local PC
The user downloads the programs and executes on the user's PC. (e.g. Windows, Linux, MacOS) There are two types, a component type and a combination type.
Combination type
- Molecular Simulation Active Workflow *This service has been terminated.
- RNA Structure Prediction Active Workflow
- Protein Structure Prediction Active Workflow*This service has been terminated.
- PhylogeneticTree (DNA, RNA, Protein) Active Workflow*This service has been terminated.
- Installation manual for KNIME and combination type active workflow
- Combination type active workflow user's manual
Developed processing node

CentroidFold_AIST: predicts RNA secondary structures.
=Ports=
in-port: connects to an out-port of each following KNIME node.
CentroidFold_AIST
=Views=
CentroidFold_AIST: displays PNG files of RNA secondary prediction result.
=Options=
Binding Site Coordinates (X, Y, Z)
There are three modes for specifying binding site coordinates.
1) Blind Docking
2) Use binding site coordinates selected by the PocketSelector node
3) Specify binding site coordinates in the input boxes
Docking Box Sizes (X, Y, Z)
There are two modes for specifying docking box sizes.
1) Use docking box sizes calculated by eBoxSize program
2) Specify docking box sizes in the input boxes
Please visit a eBoxSize web site (http://brylinski.cct.lsu.edu/content/docking-box-size) for further information.
=Ports=
in-port0(top): an absolute path of a PDB format file.
in-port1(bottom): an absolute path of a PDB format file.
out-port: an absolute path of output files.

Please visit the CentroidFold Web site (http://medals.jp/elist/detail/17.html) for further information.
=Options=
Input type: select an input type format from FASTA or ClustalW.
Output: specify an output directory for execution result file.
Weight of base pairs: select a gamma value of weight of base pairs.
Advanced: set advanced other options (optional)
=Ports=
in-port: an absolute path of an input file.
out-port: absolute paths of output files.

=Options=
Select Output Directory: Specify an absolute path of a directory for storing results of DockingAnalyzer.
=Ports=
in-port: an absolute path of a AutoDock-vina result files (PDF format).
out-port: an absolute path of output directory and receptor PDB file.

=Options=
Fasta File: set an absolute path of a FASTA file.
=Ports=
out-port: an absolute path of a FASTA file.

Please visit a fpocket2 web site (http://fpocket.sourceforge.net/) for further information.
=Options=
Select Output Directory: specify an absolute path of a directory for storing results of fpocket2_AIST.
=Ports=
in-port: an absolute path of a PDB file.
out-port: an absolute path of a directory for storing results of fpocket2_AIST.
=Options=
start residue (base) number: a start residue (base) number of a fragment PDB user need.
end residue (base) number: an end residue (base) number of a fragment PDB user need.
=Ports=
in-port: an absolute path of a PDB File.
out-port: an absolute path of a fragment PDB file.

=Ports=
in-port: An absolute path of the (result) file.
=Views=
display the (result) file as HTML.

Please visit a IPknot web site (http://medals.jp/elist/detail/154.html) for further information.
=Options=
Select Output Directory: specify an absolute path of a directory for storing results of IPknot.
-IPknot options-
-t th: threshold of base-pairing probabilities for each level
-g gamma: weight for true base-pairs equivalent to -t 1/(gamma+1)(default: -g 2 -g 4)
-e model: probabilistic model (default: McCaskill)
-r n: the number of the iterative refinement (default: 0)
-i: allow isolated base-pairs
-b: output the prediction by BPSEQ format
-P param: read the energy parameter file for the Vienna RNA package
=Ports=
in-port: an absolute path of a FASTA format file (RNA).
out-port: an absolute path of an output file.

Please visit a Jmol web site (http://jmol.sourceforge.net) for further information.
=Ports=
in-port: an absolute path of a directory storing results.
=Views=
A pop up dialog is displayed as follows:
Modeller_AIST: display model numbers and the objective function values.
MergeTargetAndLigand, InitMinMM_AIST: display model numbers and the energy scores.
RASSIE_AIST, Rascal_AIST: display model numbers.
The user can select only one radio button. After selecting radio button, the user can launch Jmol on pressing "Execute Jmol" button.
Please visit the LSDB Cross Search Web site (http://lifesciencedb.jp/dbsearch/)(Japanese version only) for further information.
=Ports=
in-port: an absolute path of the (Multi-)FASTA file.
=Views=
FASTA Header Lists: displays header lines of all sequences contained in the (Multi-)FASTA file.
LSDB Cross Search: displays a text box where can input some search words with search identifiers. By specifying search words and clicking a "LSDB cross search" button, a web browser is opened and displays search results.
Search identifiers:
AND: ' '(space) e.g. 'network socket'
OR : '|'(pipe) e.g. 'network | socket'
XOR: '!'(exclamation) e.g. 'network ! socket'
Wild Card: '*'(asterisk) e.g. 'inter*', `sphere`
Priority order: '|' > ' '(space), '!'

=Ports=
in-port: an absolute paths of a result directory (storing ligand files) and of a target data file.
out-port: an absolute paths of a result directory (storing merged files).

Input files should be located under ligand number as follows:
1/PL.crd
1/PL.pdb
1/PL.top
1/ligand.prep
2/PL.crd
2/PL.pdb
2/PL.top
2/ligand.prep
.
.
.
1: ligand number
PL.crd: protein-ligand complex coordinate file (amber format)
PL.pdb: protein-ligand complex PDB file (for a reference, optional)
PL.top: protein-ligand complex topology file (amber format)
ligand.prep: ligand prep file (amber format)
=Options=
Select Output Directory: specify an absolute path of a directory for storing results of InitMinMM_AIST.
=Ports=
in-port: an absolute path of a tar file of MmPrep result.
out-port: an absolute path of a tar file of MM result.

If the user connects Rebuild to DockingAnalyzer via flow variables ports (red ports), this program doesn't open the pop-up window because of already selecting candidates of each cluster calculated by DockingAnalyzer, and is followed by MinMM.
=Ports=
in-port: An absolute path of a directory stored Rebuild output files.
out-port: An absolute path of a directory stored Rebuild output files.

=Options=
PDB File: set an absolute path of a PDB file.
=Ports=
out-port: an absolute path of a PDB file.
=Ports=
in-port: an absolute path of a directory stored Qsite results.
out-port: an absolute path of a directory stored Qsite results.

Please visit a RactIP web site (http://medals.jp/elist/detail/153.html) for further information.
=Options=
Select Output Directory: specify an absolute path of a directory for storing results of RactIP.
-RactIP options-
-p: do not use the constraints for interenal pseudoknots.
-a alpha: weight for hybridation probabilities (default: 0.5).
-t th_bp: threshold of base-pairing probabilities (default: 0.5).
-u th_hy: threshold of hybridazation probabilities (default: 0.2).
-m: use McCaskill model (default: CONTRAfold model).
-i: allow isolated base-pairs.
=Ports=
in-port0(top): an absolute path of a single FASTA format file (RNA).
in-port1(bottom): an absolute path of a single FASTA format file (RNA).
out-port: an absolute path of an output file.
=Options=
Select Output Directory: specify an absolute path of a directory for storing results of Rascal.
=Ports=
in-port: an absolute path of a RactIP result file.
out-port: an absolute path of a Rascal result directory.
=Options=
Select Output Directory: specify an output directory for execution result file.
-RASSIE options-
-q Nstruct
-ins insertion_num
-clst -outclst n
-ins_chain
=Ports=
in-port: an absolute path of a result file of RNA secondary structure prediction.
out-port: absolute paths of RASSIE result files.
=Options=
Select Output Directory: specify an absolute path of a directory for storing results of Rebuild.
=Ports=
in-port: an absolute path of a AutoDock-vina result files (PDF format).
out-port: an absolute path of output directory.
=Ports=
in-port: an absolute path of a result file of RNA secondary structure prediction.
out-port: an absolute path of a result file of RNA secondary structure prediction.
=Ports=
in-port: an absolute path of a SPARQL result file.
out-port: an absolute path of selected sequence FASTA-format file.

=Dialog Options=
Active flow variable output port number:
0: first flow variable output port
1: second flow variable output port
2: third flow variable output port

The user can also input SPARQL sentence in "Input SPARQL Query" text area. If the user input the SPARQL sentence, other options are entirely-ignored except for "Output directory" option.
=Options=
Output directory: specify an absolute path of directory to store SPARQL results.
Sparql endpoints: specify SPARQL endpoints.
Species name: specify (a) species name(s) as search parameters.
Keyword: specify (a) keyword(s) as search parameters (not available for UNIPROT).
Minimum sequence length: specify a minimum sequence length thresholds as search parameters.
Maximum sequence length: specify a maximum sequence length thresholds as search parameters.
Resolution: specify a Resolution (for PDB) as search parameters.
Pathway: specify a pathway (for KEGG-pathway) as a search parameter.
Output format: specify either FASTA or Tab-delimited.
Advanced: input SPARQL sentence.
=Ports=
out-port: an absolute path of a FASTA-format or Tab-delimited output file.
=Options=
Output directory: specify an absolute path of directory to store SPARQL results.
Endpoint: specify an endpoint.
Advanced: input SPARQL sentence.
=Ports=
out-port: Specify an absolute path of a Tab-delimited output file.
=Messege=
REST execution error. Please check your input file. If you have any questions, please let us know (workflow@medals.jp).
=Nodes=
All nodes executed via REST.
=Messege=
Sorry, system is busy. Please try later.
=Nodes=
All nodes executed via REST.
=Messege=
Time out error has occured. "program name" program failed in calculating in time.
=Nodes=
All nodes executed asynchronously via REST.
=Messege=
The total file size is "total size" bytes. Maximum total size is 32MB.
=Nodes=
All nodes executed via REST.
=Messege=
A Multi-FASTA file is not permitted. Please input a single FASTA file.
=Nodes=
All nodes that only permit a single FASTA format as input.
=Messege=
Sequence length limit is "sequence length"aa. Please input more short sequence.
=Nodes=
All nodes that have an allowed length of sequence.
=Messege=
Your setting file does not exist.
=Nodes=
AlignmentFileReader, FastaFileReader, Mol2FileReader and PdbFileReader nodes.
=Messege=
No results found. Please change your query conditions.
=Nodes=
CompoundQuery_AIST node.
=Messege=
SPARQL RESULTS: 0 data hits. No hit. Please change your search conditions.
=Nodes=
All Sparql node.
=Messege=
Plase check SPARQL endpoints.
=Nodes=
All Sparql node.
REST/REST services
The user executes programs on servers at AIST and receives the results using REST interface. (Internet connection is required. )